Modelo espacial vs Modelos clásicos
Una duda que podría surgirnos en el momento de analizar nuestros ensayos de campo es qué tan bueno puede ser un modelo que tenga en cuenta componentes espaciales, frente a un diseño experimental tradicional como lo es un diseño completamente al azar (CRD), un diseño en bloques completos al azar (RCBD) o un diseño alfa lattice . Definimos aquí el alfa lattice como un diseño en bloques completos (todos los genotipos dentro de cada bloque) y dentro de cada bloque o repetición, bloques incompletos compuestos por un subconjunto de genotipos.
Ejemplo con datos de fríjol
Como ejemplo se tomarán datos de Darien en el añó 2016 bajo condiciones de sequía. En este se evaluaron un total de 380 lineas en dos repeticiones, con 18 bloques incompletos dentro de cada una.
| YdHa | rep | block | col | row | line | PLHA | col_f | row_f |
|---|---|---|---|---|---|---|---|---|
| 507.0637 | R1 | B1 | 1 | 1 | DAB_913 | 20 | 1 | 1 |
| 1077.5100 | R1 | B1 | 1 | 2 | RAA_021 | 24 | 1 | 2 |
| 802.8509 | R1 | B1 | 1 | 3 | CAL_143 | 30 | 1 | 3 |
| 633.8297 | R1 | B1 | 1 | 4 | ACC_004 | 23 | 1 | 4 |
| 528.1914 | R1 | B1 | 1 | 5 | DAB_267 | 20 | 1 | 5 |
| 591.5744 | R1 | B1 | 1 | 6 | DAN_003 | 25 | 1 | 6 |
Con el objetivo de completar el campo algunos genotipos (AFR_298, AFR_722, CAL_096, CAL_143, DAA_153, DAB_251, DAB_295, DAB_494, DAB_614, DAB_932, RAA_034, SAB_560, SAB_659, SAB_686, SEQ_1003, TIO_CANELA_75), fueron replicados 4 veces, completando así un total de 792 observaciones.
Figure 1: Blocks in the field
Ahora bien, se ajustará primero los modelos tradicionales haciendo uso de la librería lme4 y los modelo espaciales haciendo uso de SpATS y asreml. A continuación se muestra la sintaxis necesaria para ajustar los modelos.
Datos$blo_in_rep <- with(Datos,rep:block)
# Completely Randomized Designs (CRD)
g.ran <- lmer(data = Datos , formula = YdHa ~ 1 + (1|line), REML = T )
# Randomized Complete Block Designs (RCBD)
g.ran2 <- lmer(data = Datos , formula = YdHa ~ 1 + (1|line) + rep, REML = T )
# Alpha-lattice
g.ran3 <- lmer(data = Datos , formula = YdHa ~ 1 + (1|line) + rep+ (1|blo_in_rep), REML = T )# SpATS
Modelo=SpATS(response='YdHa',
genotype='line', genotype.as.random=T,
fixed=NULL ,
spatial = ~ PSANOVA(col, row, nseg = c(ncols,nrows), degree=c(3,3),nest.div=2),
random = ~ row_f + col_f , data=Datos,
control = list(tolerance=1e-03, monitoring=0))Datos <- arrange(Datos,col,row)
# Filas anidadas en las columnas
fit.asreml<- asreml( fixed = YdHa ~ 1 + lin(col) ,
random = ~ line + row_f + spl(row) + spl(col)+ units ,
data = Datos,
rcov = ~ ar1(col_f):ar1(row_f),
maxiter = 100,
trace = FALSE, na.method.X = "include", na.method.Y = "include")Como medidas de comparación se hará uso de los criterio de información (AIC, BIC), el r2, la heredabilidad y finalmente la desviación estándar (SD) residual de los modelos.
| Models | aic | bic | r.2 | heri | var.r |
|---|---|---|---|---|---|
| DCA | 11709.842 | 11723.851 | 0.406 | 0.416 | 358.73 |
| RCBD | 11689.588 | 11708.266 | 0.431 | 0.434 | 353.10 |
| Alpha | 11509.962 | 11533.309 | 0.680 | 0.542 | 281.91 |
| SpATS | 9865.164 | 9925.868 | 0.824 | 0.650 | 225.38 |
| ASreml | 9890.131 | 9927.466 | 0.929 | 0.657 | 157.85 |
La tabla 2 muestra cómo bajo todos los criterios se obtienen mejores resultados en los modelos espaciales. El AIC y el BIC fueron más bajos en SpATS, el r2 y la Heredabilidad aumentaron aproximadamente en un 42% y 23%, respectivamente, y la desviación estándar residual logró reducirse considerablemente en los dos últimos modelos. Cabe resaltar que la desviación estándar residual de ASReml, que se muestra en la tabla 2 se encuentra sin el efecto nugget que tuvo una SD de 213.09.
Finalmente, de manera gráfica miraremos qué sucede espacialmente cuando se ajusta cada uno de los diseños, finalizando con la matriz de correlaciones de los efectos genéticos entre modelos.
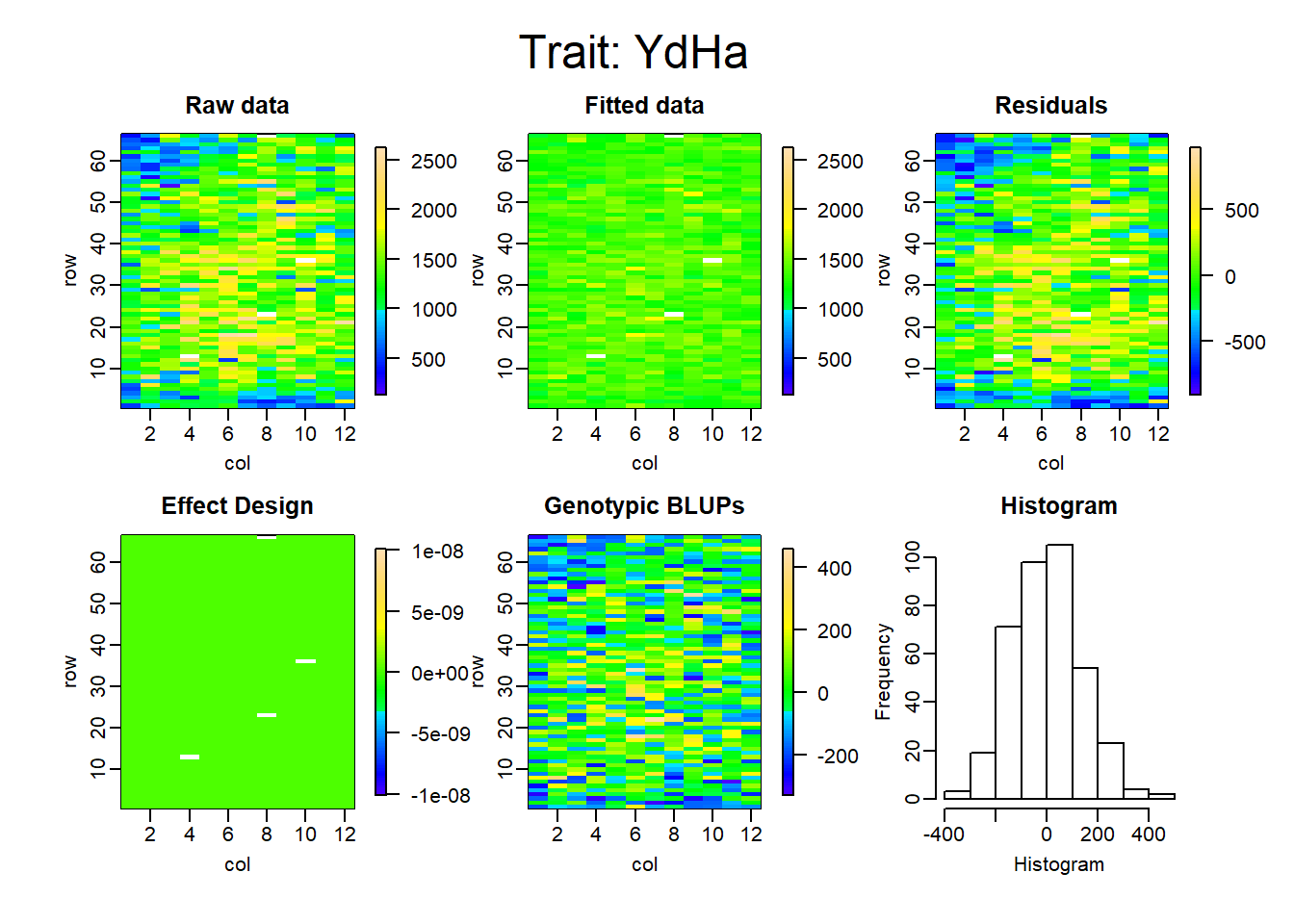
Figure 2: Completely Randomized Designs (CRD)
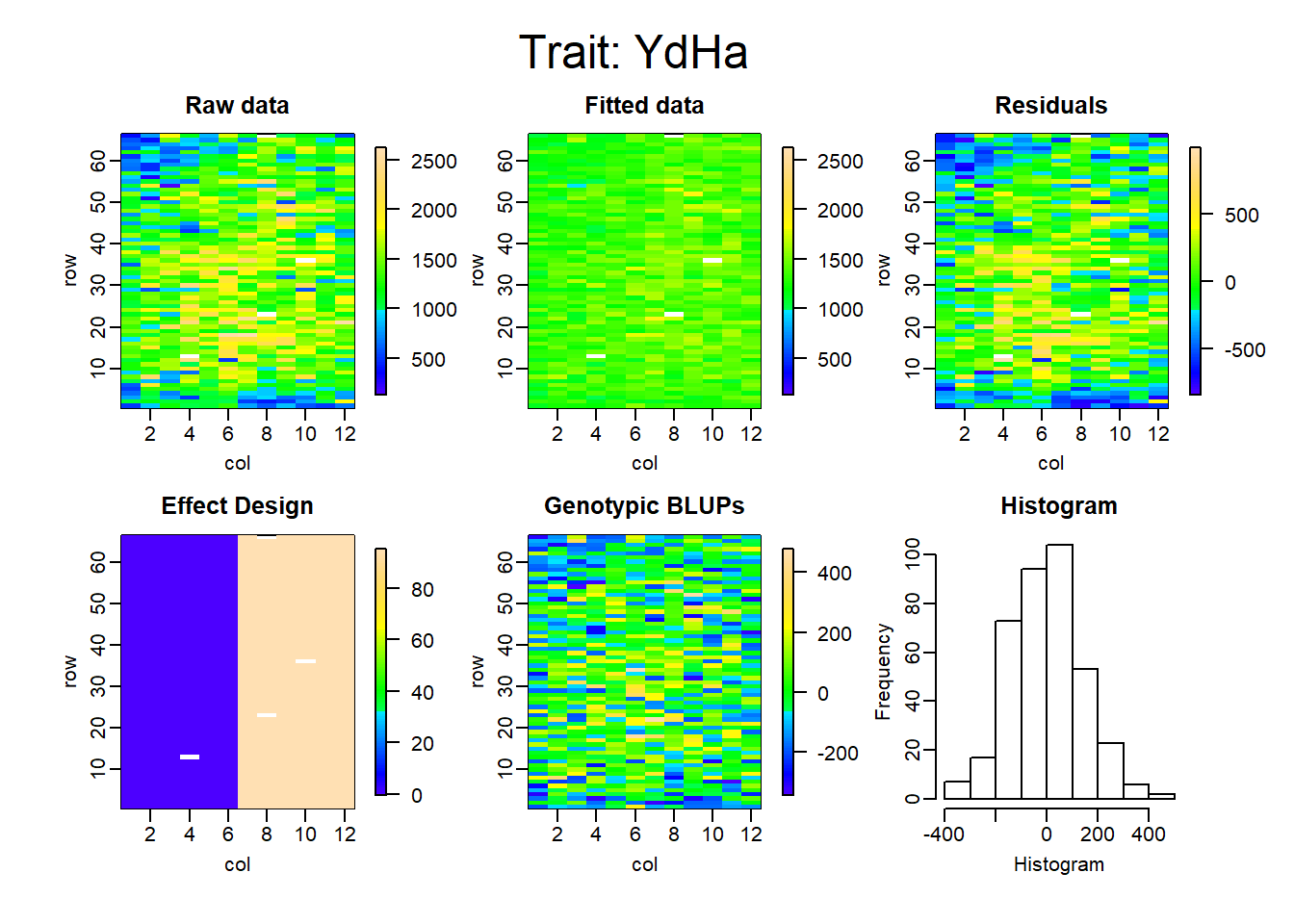
Figure 3: Randomized Complete Block Designs (RCBD)
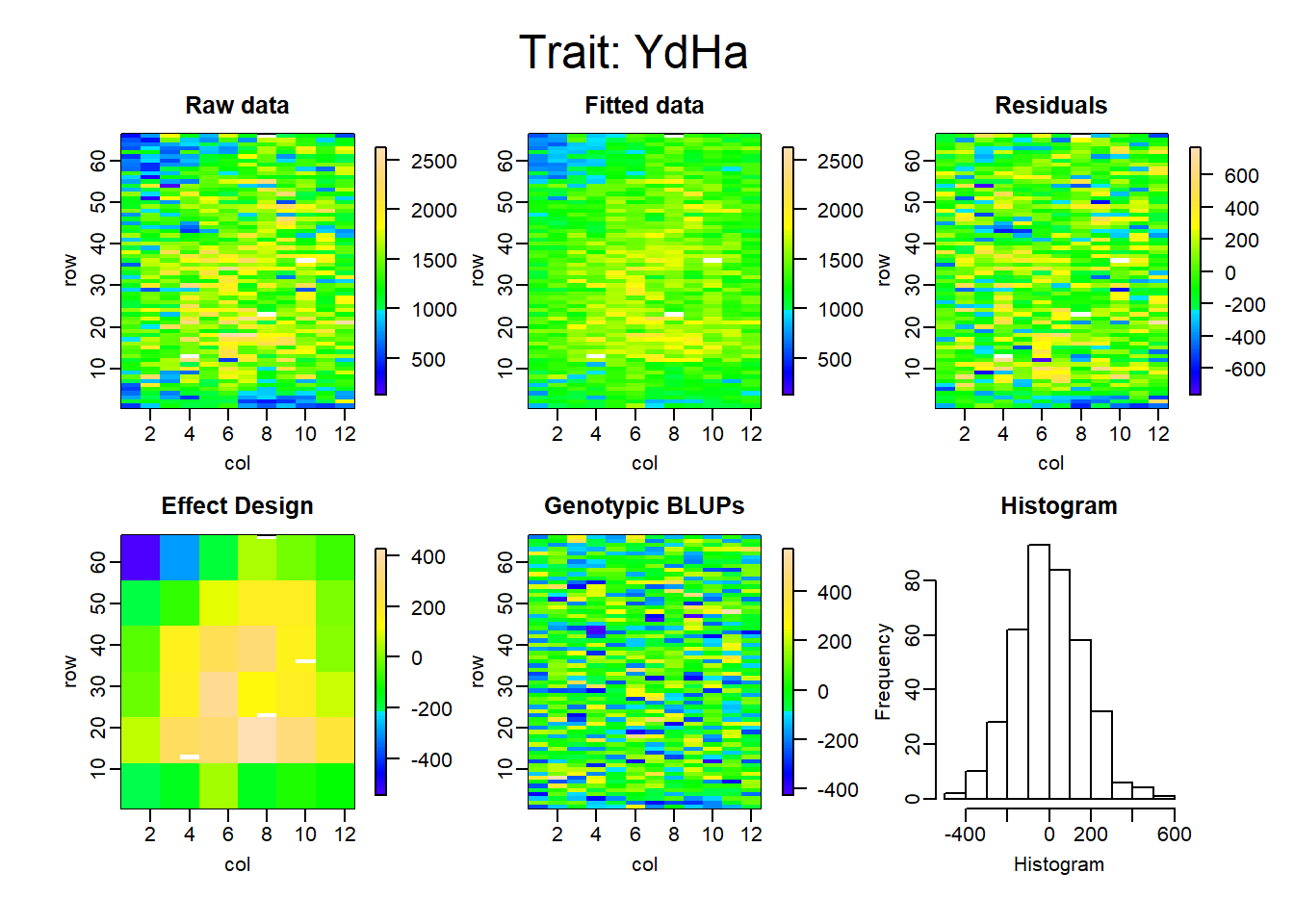
Figure 4: Alpha-Lattice Design
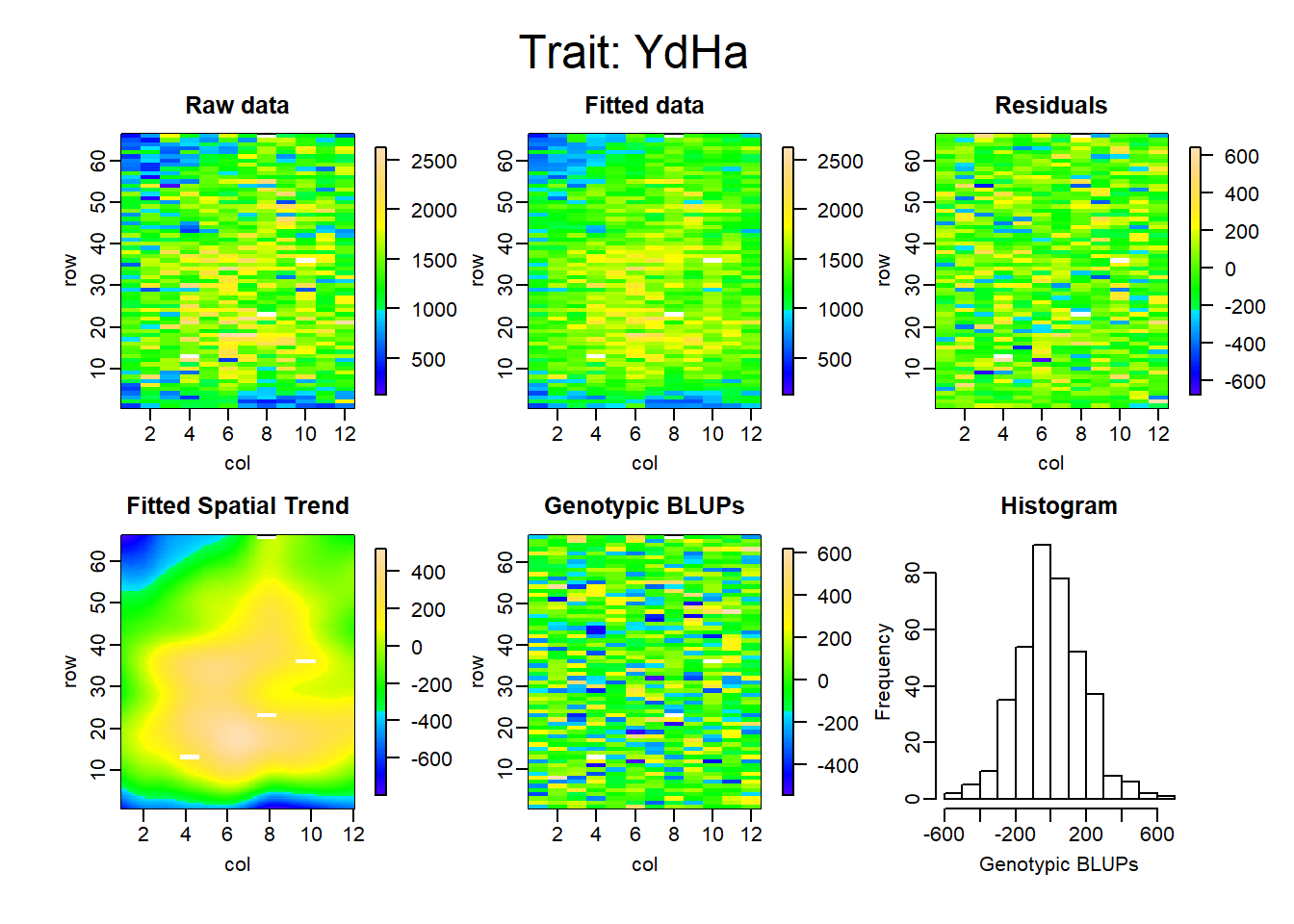
Figure 5: Spatial model with SpATS
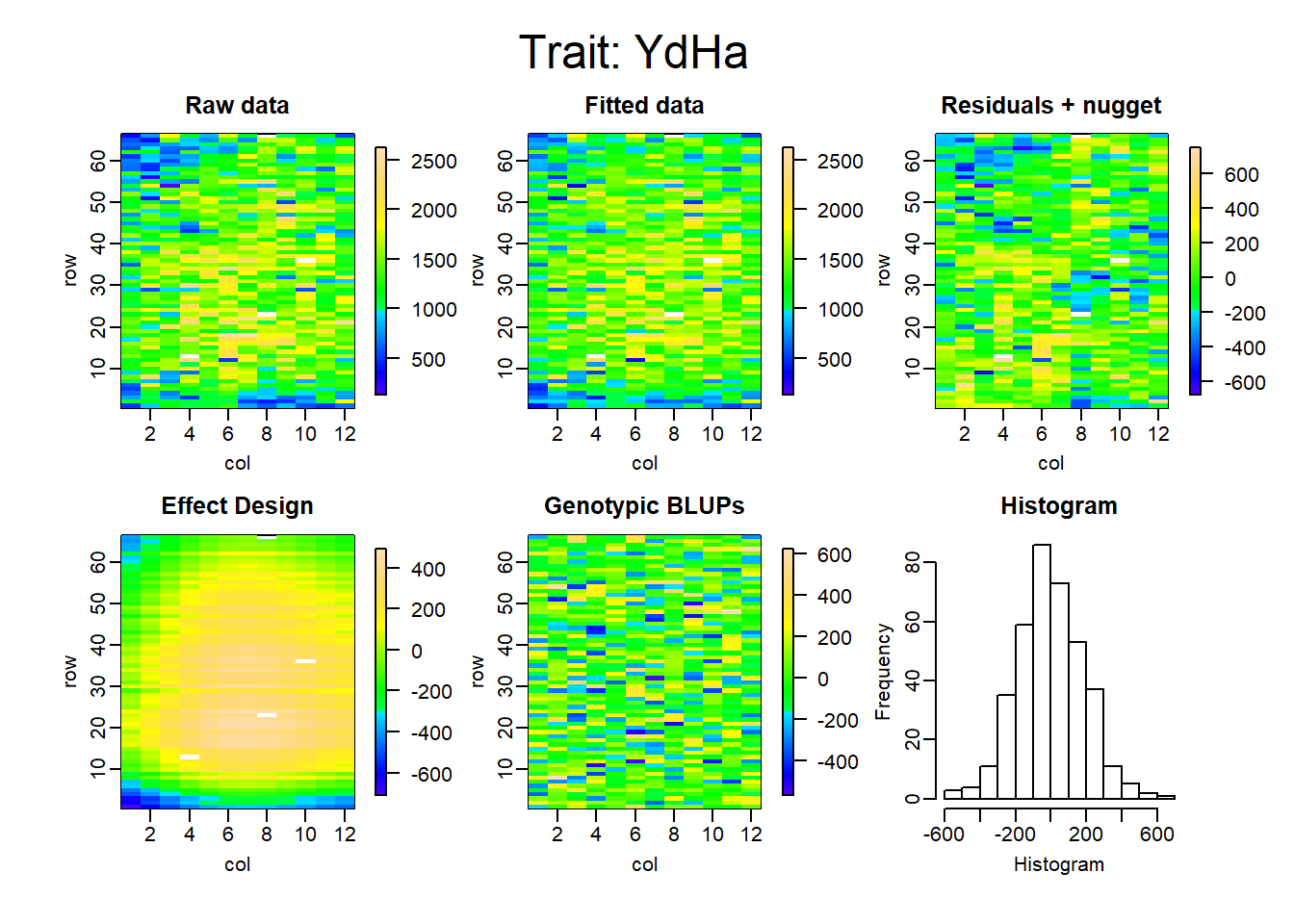
Figure 6: Spatial model with ASReml
Las correlaciones entre los BLUPs de los modelos propuestos vendrían dadas por:
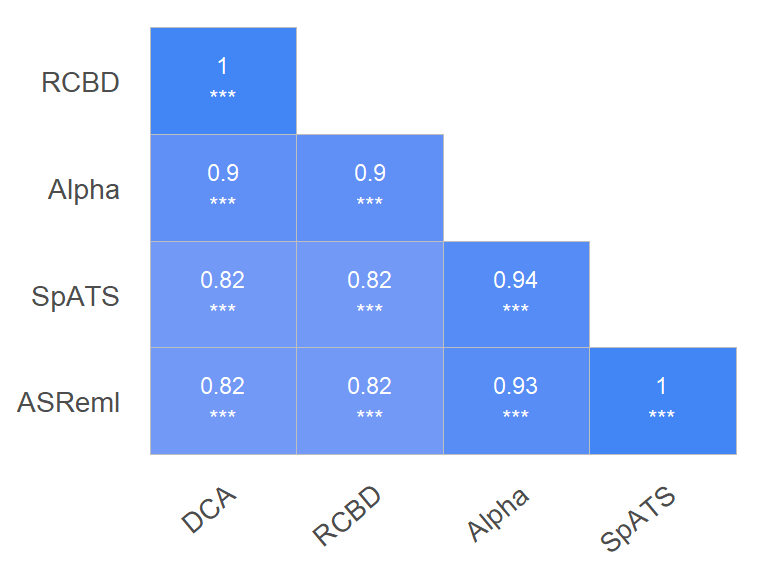
Figure 7: Correlations Matriz
Algunos de los códigos que no se presentaron aquí, se pueden encontrar en el GitHub:
Ejemplo con datos de trigo
Como ejemplo se tomará un experimento de trigo bajo un diseñado en bloques completos al azar (RCBD) en el sur de Australia (Gilmour et al., 1997).
| yield | geno | rep | row | col | rowcode | colcode |
|---|---|---|---|---|---|---|
| 483 | 4 | 1 | 1 | 1 | 2 | 1 |
| 526 | 10 | 1 | 2 | 1 | 2 | 1 |
| 557 | 17 | 1 | 3 | 1 | 1 | 1 |
| 564 | 16 | 1 | 4 | 1 | 2 | 1 |
| 498 | 21 | 1 | 5 | 1 | 2 | 1 |
| 510 | 32 | 1 | 6 | 1 | 1 | 1 |
En este experimento un total de 107 variedades de trigo fueron evaluadas en 3 bloques completos, donde cada bloque consistió de 5 columnas y 22 filas en el campo (3 variedades se probaron 2 veces en cada réplica). Completando así un total de 330 surcos.
RCBD
Se hará uso de la librería lme4 para ajustar el modelo RCBD:
# RCBD
Model.lme4.1 <- lmer(yield ~ 1 + (1|geno) + rep , data=wheatdata , REML = T)
# Modelo con factores asociados a la siembra y a la cosecha
Model.lme4.2 <- lmer(yield ~ 1+(1|geno)+rep + colcode+rowcode , data=wheatdata , REML = T)En este se especifica, primero, la variable respuesta, seguidamente del intercepto o media global (opcional), el genotipo como factor aleatorio y finalmente la repetición, colcode y rowcode, como factores fijos. El metodo de estimación se especifica como Maxima verosimilitud restringida (REML).
Corrección espacial
Ahora bien, ajustaremos el modelo con corrección espacial haciendo uso de la librería SpATS, la sintaxis necesaria se describe a continuación:
wheatdata$col_f = factor(wheatdata$col)
wheatdata$row_f = factor(wheatdata$row)
ncols = length(unique(wheatdata$col))
nrows = length(unique(wheatdata$row))Con lo anterior se definen las coordenadas fila/columna como factores y el número de segmentos o nodos para el ajuste de los suavizadores Spline.
Model.spats.1 <-SpATS(response='yield',
genotype='geno', genotype.as.random=T,
fixed=NULL ,
spatial = ~ PSANOVA(col, row, nseg = c(ncols,nrows), degree=c(3,3),nest.div=2),
random = ~ row_f + col_f , data=wheatdata,
control = list(tolerance=1e-03, monitoring=0))
# Modelo con factores asociados a la siembra y a la cosecha
Model.spats.2 <-SpATS(response='yield',
genotype='geno', genotype.as.random=T,
fixed= ~ colcode + rowcode ,
spatial = ~ PSANOVA(col, row, nseg = c(ncols,nrows), degree=c(3,3),nest.div=2),
random = ~ row_f + col_f , data=wheatdata,
control = list(tolerance=1e-03, monitoring=0))Resultados
Los gráficos que se muestran a continuación representan la descomposición de las componentes para cada uno de los modelos ajustados.
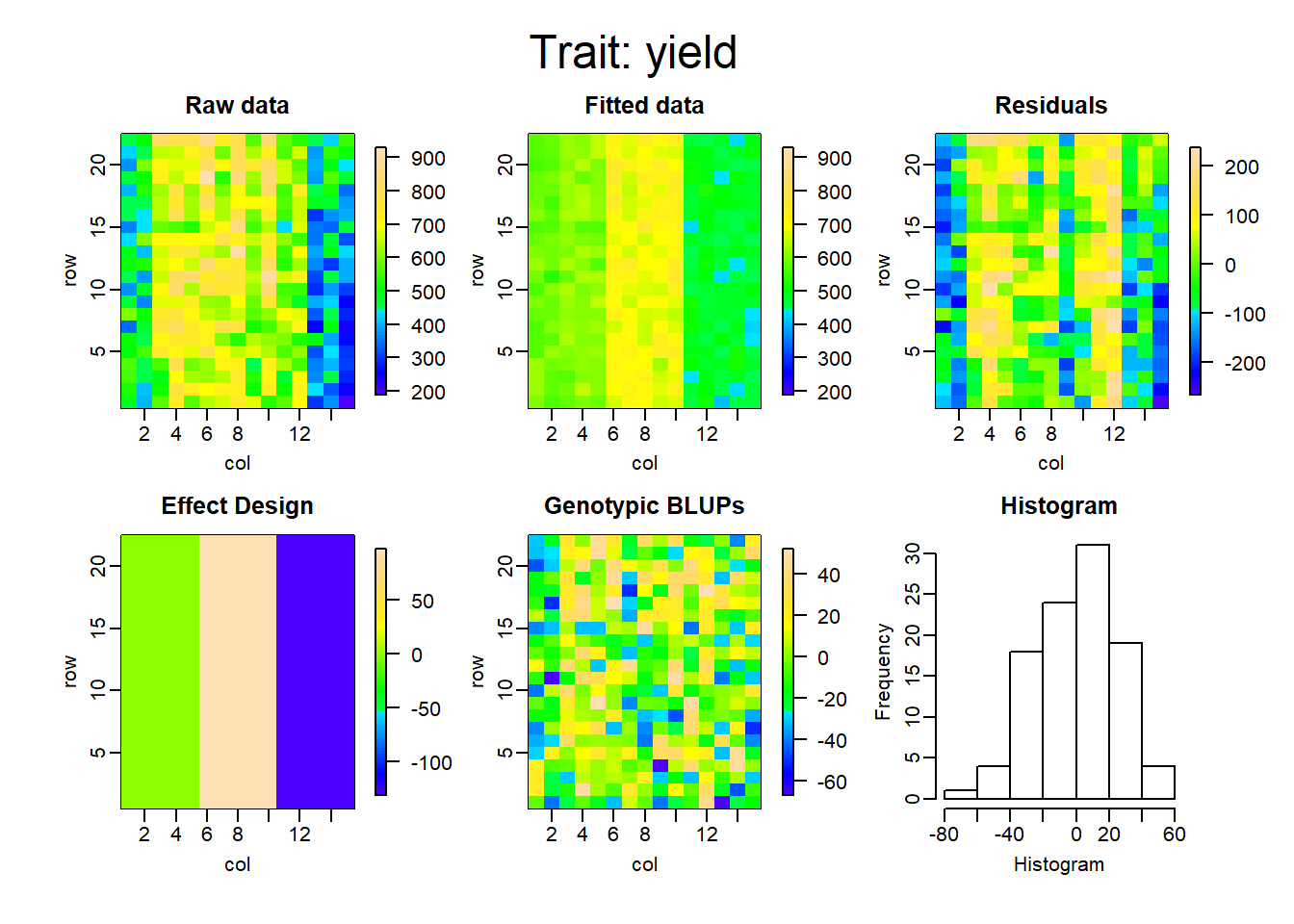
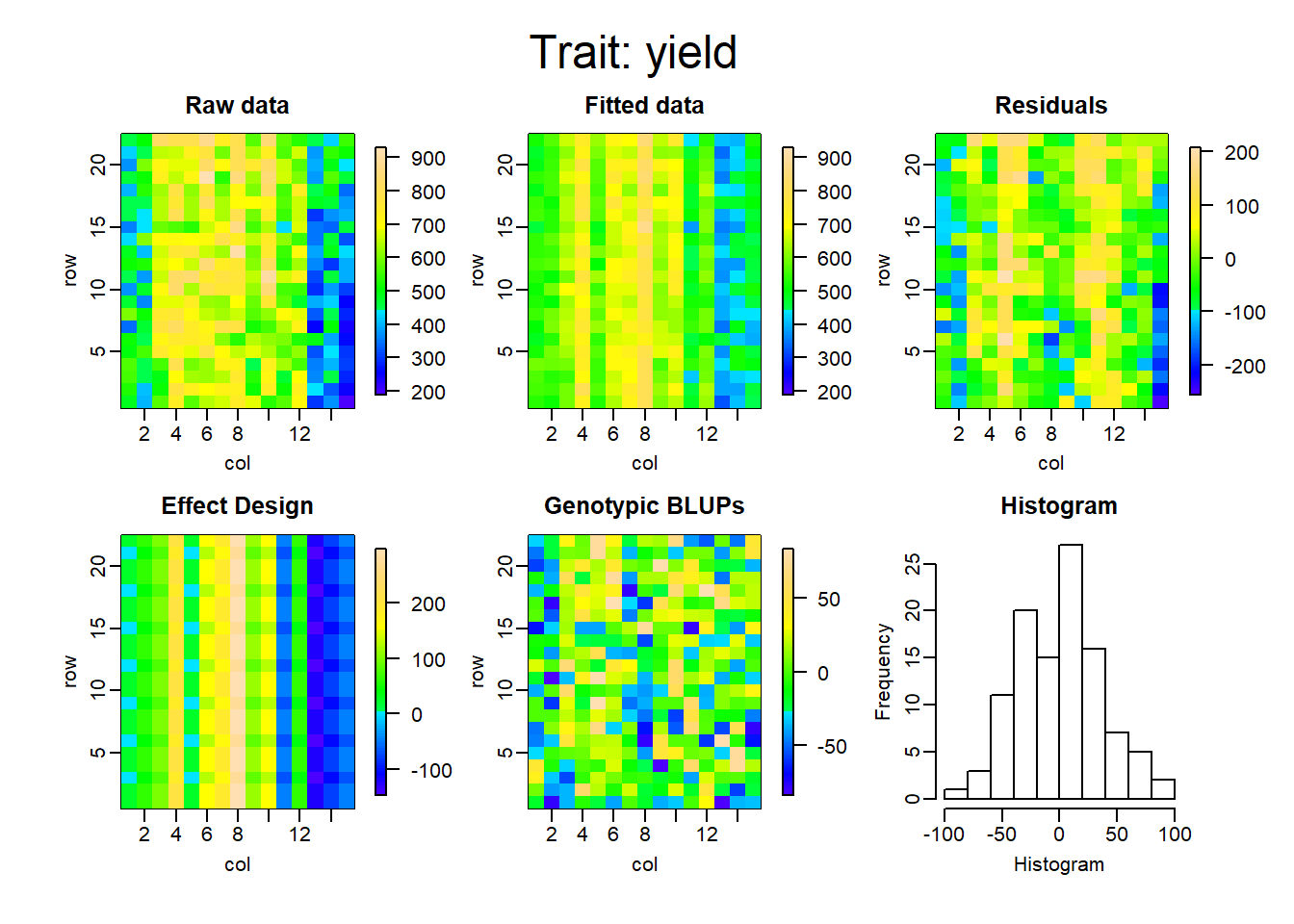
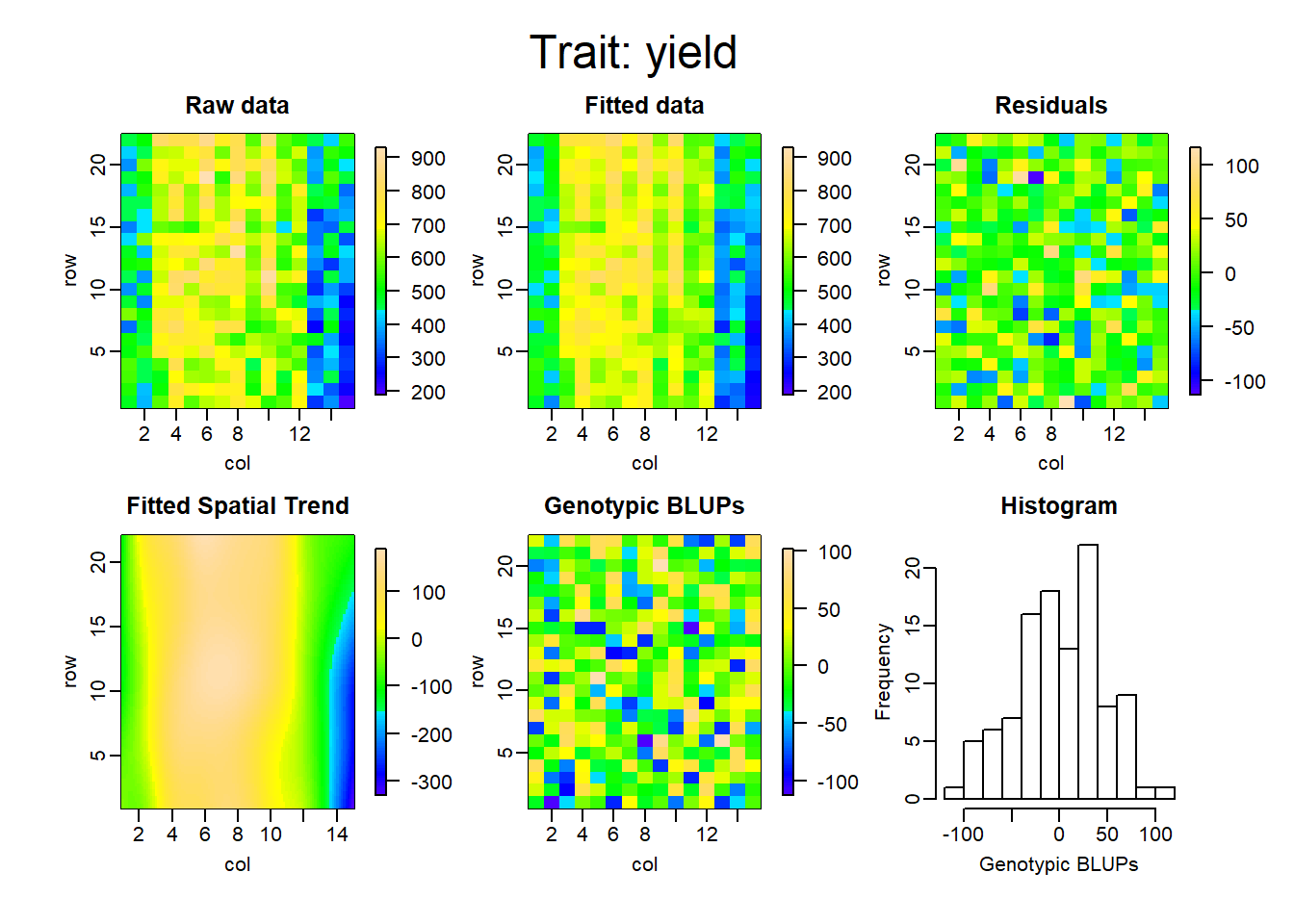
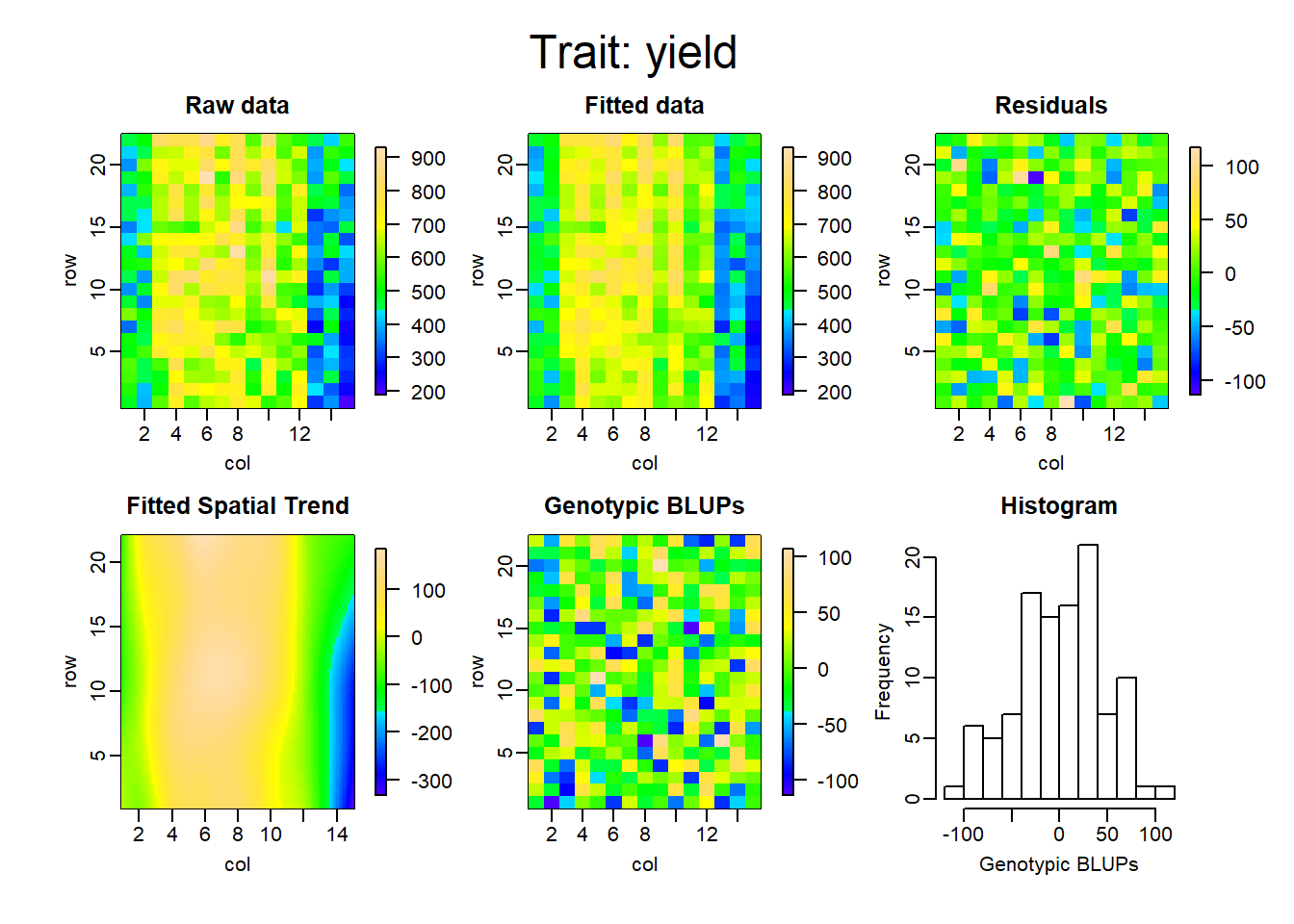
La tabla a continuación muestra la comparación de ambas metodologías, en cuanto al criterio de información de akaike (AIC), el R cuadrado, la varianza residual del modelo y finalmente la heredabilidad.
| aic.lme4 | aic.SpATS | r.lme4 | r.spats | res.lme4 | res.spats | h.lme4 | h.spats |
|---|---|---|---|---|---|---|---|
| 4096.927 | 3109.310 | 0.498 | 0.949 | 115.45 | 44.40 | 0.31 | 0.77 |
| 3960.924 | 3071.424 | 0.702 | 0.949 | 92.62 | 44.15 | 0.49 | 0.77 |
De esta manera, es posible ver cuanto mejora el ajuste del modelo teniendo en cuenta el arreglo espacial. El r cuadrado, en el primer modelo, pasó de 0.49 a 0.95, comparando RCBD con SpATS. Lo que equivale aproximadamente a un aumento del 45% en el porcentaje de variabilidad explicado por el modelo. Por otro lado, la varianza residual logra reducirse considerablemente y se logra también un aumento en la heredabilidad en un 46%.
Finalmente vemos cómo el incorporar los efectos adicionales de la siembra y la cosecha, disminuyen el AIC y la varianza residual, y aumentan el porcentaje de variabilidad explicado y la heredabilidad del modelo.